Abstract
Creutzfeldt–Jakob disease (CJD) is a fatal disease caused by the accumulation of abnormal prion proteins in neurological tissues. Routine notification data reveal that NSW has similar rates of CJD to other states and territories in Australia; however, it is likely that there is significant under-ascertainment of cases. It is important that clinicians and public health staff remain vigilant for the clinical signs of CJD and understand the limitations of the different diagnostic tests available. This paper provides a brief overview of the epidemiology of CJD in NSW, as well as current issues in the diagnosis and public health investigation of CJD.
Full text
Introduction
Creutzfeldt–Jakob disease (CJD) is a type of transmissible spongiform encephalopathy caused by replication of abnormal prion proteins, mainly in neurological tissues. It is typically characterised by spongiform pathology of the brain.1 Prion proteins are normally present in all cells and are likely to play a role in cell adhesion and intracellular signalling. However, their exact nature and role in the pathogenesis of CJD is still debated.1,2 The normal prion protein (PrPc) becomes abnormal through conformational change and is then typically resistant to proteases because of its β-sheet structure.3,4 The accumulation of abnormal prion protein (PrPsc) results in neuronal cell death, gliosis and microglial activation in neurological tissue.5 PrPsc can spontaneously arise in tissues (sporadic CJD; sCJD), be produced from a mutation in the prion protein gene (PRNP; genetic CJD) or be transmitted via infected tissues in different ways (iatrogenic CJD). Table 1 summarises the prevalence and clinical course of different types of CJD. It is important to note that, irrespective of the type of disease, the average time between onset of symptoms and death is less than 12 months in Australia. The Australian National CJD Registry (ANCJDR) was set up in 1993, following four medically acquired CJD deaths in Australia.6 Since then, the ANCJDR surveillance program has increased in scope to prospectively ascertain and investigate all cases of CJD, and the register has attempted to retrospectively obtain data on all cases since 1970.7 Data from the registry reveal that, between 1970 and 2012, 90% of the 482 confirmed cases of CJD in Australia were classified as sCJD.7 In Australia, the average annual age-adjusted mortality rate for CJD of 1.2 deaths per million population is similar to that previously reported across Europe and Canada.7,8 The aim of this paper is to update public health practitioners on the epidemiology of CJD in NSW and provide an overview of the key issues associated with the investigation and public health management of CJD cases.
Surveillance and epidemiology of CJD in NSW
Surveillance
In NSW, all possible, probable and confirmed cases of ‘classical’ CJD are notified by medical practitioners and/or laboratories to public health unit (PHU) staff9 (see Box 1). PHU staff ensure that the case is notified to the ANCJDR and contribute to the local investigation to identify potential risk factors and infection control issues.9 Only probable and confirmed cases are entered into the NSW Notifiable Conditions Information Management System (NCIMS). A probable case requires clinical evidence, including a progressive dementia of less than two years and either a typical electroencephalogram (EEG) or positive 14-3-3 protein in cerebrospinal fluid (CSF) (Box 1). A confirmed case requires identification of a progressive neurological disorder and histopathological confirmation, typically performed on a limited autopsy of the brain postmortem (Box 1).
Epidemiology
To examine trends in NSW CJD cases over time, data were extracted from NCIMS for all classical CJD cases (this includes all probable and confirmed cases of sporadic, genetic and iatrogenic CJD) from 2004 to 2013, with a total of 83 CJD cases reported. Age-standardised rates per 100 000 population were calculated using estimated resident midyear population data from the Australian Bureau of Statistics for the same period. Four cases were excluded from further analysis because the NSW resident location was not recorded or the case did not reside within one of the NSW Health Local Health Districts (LHDs) (i.e. was a resident of the Network with Victoria boundary). Of the remaining 79 classical cases, 48 were confirmed as CJD on histopathological examination of brain tissue.
Table 1. Types of Creutzfeldt–Jakob disease (CJD) in Australia
| Type | Prevalence | Transmission/cause | Median age at death | Median disease durationa |
|---|---|---|---|---|
| Classical Genetic |
~7% of all cases | Dominantly inherited point mutations in the PRNP gene on chromosome 20 | 59 years (range: 18–82 years)7 |
6 months (range: 1.5–192 months)7 |
| Classical Sporadic |
~90% of all cases | Presumed spontaneous conversion of PrPc to PrPsc | 66 years (range: 25–90 years)7 |
3.5 months (range: 0.9–60 months)7 |
| Classical and acquired Iatrogenic |
Eight confirmed cases reported since 1970 | Contaminated tissues infected with PrPsc Five cases due to Lyodura grafts and three cases due to pituitary-derived human gonadotropin hormone replacement therapy |
39 years (range: 26–62 years)10 |
6.5 months (range: 2–25 months)10 |
| Acquired variant CJD |
No known cases reported | Infection with PrPsc of bovine origin | 28 years11 | 14 months11 |
PRNP = prion protein gene; PrPc = normal prion protein; PrPsc = abnormal prion protein
Disease duration = time from onset of symptoms to death.
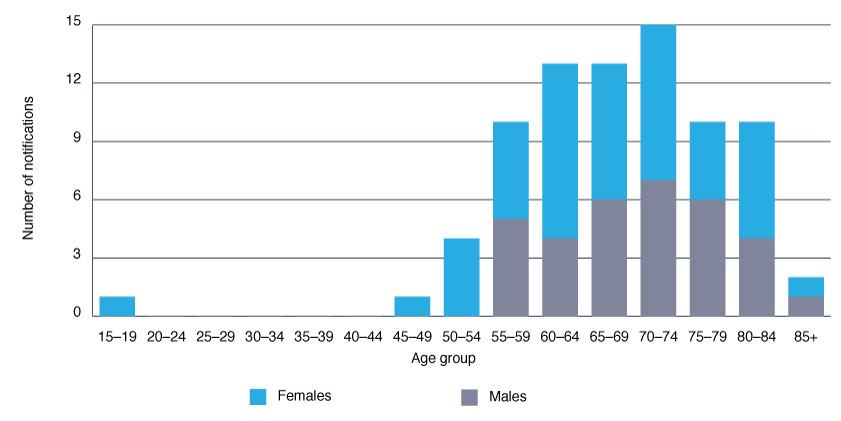
Each year, 6–10 cases have been notified in NSW (Figure 1), with a stable age-standardised rate of around one case per million population per year, similar to the reported Australian7 and worldwide incidence rate of CJD.8 The disease predominantly affects older age groups, and slightly more females than males are diagnosed (Figure 2), reflecting Australian data published by the ANCJDR.7
Age-standardised rates of classical CJD across LHDs in NSW varied between 2004 and 2013 (Figure 3), ranging from 0.26 (Mid North Coast LHD) to 2.1 (Illawarra Shoalhaven LHD) per million population, with no statistically significant differences between the LHD rates and all NSW. The average rate of CJD notification across NSW was 0.98 per million population (95% CI 0.76–1.19) for 2004–13. Klug et al.12 previously reported a cluster of sCJD (n = 14) in northern coastal NSW between 1993 and 2006. Further investigation of the cluster revealed that clinicians in this region were significantly more likely to refer suspect CJD cases for diagnostic investigation than in other regions, and sustained a higher level of clinical vigilance over time, leading to increased detection of CJD in the region.12
Klug et al.12 suggested that the true incidence of sCJD may be higher than currently indicated by routine surveillance data and more in line with recent autopsy studies from Europe (i.e. rates up to 2.37 cases per million population).13 A similar relationship between surveillance intensity and disease incidence has also been reported in other countries.14
Box 1. Case definition and criteria for Creutzfeld-Jakob disease (CJD) in NSW
Reporting CJD: ‘classical’ cases (including sporadic, genetic and iatrogenic forms)
Possible case:
- Progressive dementia of less than two years duration, and
- Electroencephalogram (EEG) atypical or not known, and
- At least two of the following clinical features
- Myoclonus
- Visual or cerebellar disturbance
- Pyramidal/extrapyramidal dysfunction.
Probable case:
- Progressive dementia of less than two years duration, and
- Typical EEG (e.g. presence of periodic, 1-2 Hz sharp wave discharges), and/or positive 14-3-3 protein in cerebrospinal fluid
- At least two of the following clinical features
- Myoclonus
- Visual or cerebellar signs
- Pyramidal or extrapyramidal signs
- Akinetic mutism.
Confirmed case:
- Progressive neurological disorder, and
- Neuropathological confirmation on examination of brain tissue, supplemented by immunochemistry for protease-resistant PrPsc (western blot).
Source: NSW Ministry of Health case definition, www.health.nsw.gov.au/Infectious/controlguideline/Pages/cjd.aspx
Key issues associated with public health investigation of CJD cases
Clinical signs and symptoms
Clinical signs and symptoms of CJD vary according to the molecular subtype of the disease, which is in part due to a polymorphism (coding for methionine or valine) in codon 129 of PRNP.2,4 Sporadic CJD has two main phenotypic subtypes: cognitive sCJD and ataxic sCJD. Cognitive sCJD accounts for 55–70% of all sporadic cases. These cases are fairly typical in presentation, with rapidly progressive cognitive impairment and confusion, followed by cortical visual disturbances, ataxia and/or myoclonus.2 Ataxic subtypes commonly present with ataxia, with or without cognitive impairment.2
Figure 1. Notifications and age-standardised rates for probable and confirmed classical Creutzfeldt–Jakob disease in NSW residents, 2004–13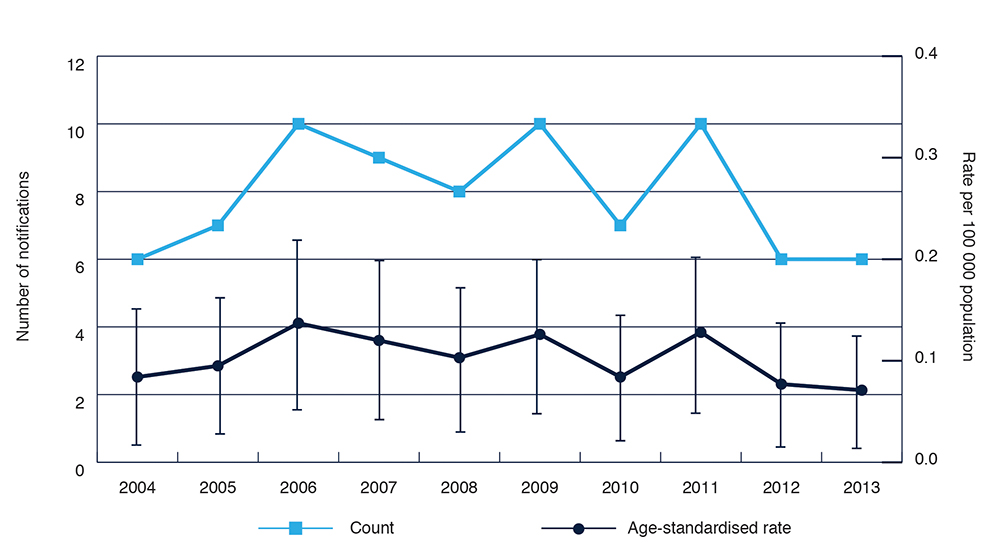
Note: Error bars show 95% confidence intervals
Figure 2. Age and sex distribution of total notifications for probable and confirmed classical Creutzfeldt–Jakob disease in NSW residents, 2004─13
Diagnosis
Confirmation of a CJD diagnosis requires neuropathological examination, which usually involves a limited autopsy of the brain postmortem with histopathological examination of fixed brain tissue, including immunohistochemistry for prion protein deposits. Brain biopsy is not recommended on living patients for the diagnosis of CJD; however, it may be deemed appropriate to rule out certain alternative diagnoses.9 As the changes of CJD are not uniformly distributed throughout the brain15, neurosurgical biopsies should be targeted at regions showing the most significant magnetic resonance imaging (MRI) changes, to reduce the chance of false negative results.
Three main antemortem tests are used to diagnose CJD: an EEG16 to detect periodic complexes in the brain, MRI of the brain15 to detect characteristic lesions and testing for the 14-3-3 protein in CSF17 (see Table 2). The ANCJDR can also facilitate genetic testing and counselling for individuals and families with suspected genetic CJD.
These tests are used in combination with clinical criteria to identify probable cases of CJD (Box 1). However, the 14-3-3 protein is a nonspecific marker of central nervous system damage and may also be positive in conditions such as recent stroke, encephalitis and other dementing disorders.22 From 2002 to 2008, a review of the performance of the 14-3-3 test revealed a sensitivity and specificity of approximately 88% and a positive predictive value of 64% (G Klug, ANCJDR, pers. comm., September 2013). These figures are based on cases where a clinical outcome has been ascertained by the ANCJDR after either follow-up investigation (38% of all 14-3-3 test referrals) or neuropathological examination (11% of all 14-3-3 test referrals). Evidence from the US17 and Australia23 indicates that the accuracy of the 14-3-3 test in diagnosing CJD is heavily dependent on the pre-test probability of CJD. This means that increased awareness and understanding of the presenting clinical signs will potentially improve the accuracy of CJD diagnosis.
ANCJDR data reveal that 62% of suspect notified cases where death is known to have occurred undergo an autopsy.7From 1993 to 2011, 46% of suspect cases notified to the registry were subsequently classified as either definite CJD (i.e. via postmortem histopathological examination) or probable (by clinical evaluation by the ANCJDR after death). Definite and probable cases are reported in incidence data.7
A recent review of notification and neuropathological examination data from CJD registries across Australia, the UK and Europe reveals lower annual average referral rates for neuropathological confirmation of suspect cases (0.88–2.64 per million examinations per year) compared with CSF 14-3-3 protein testing referrals (0.92–17.25 per million examinations per year).14 Autopsy is a sensitive issue to discuss and requires family consent. However, the ANCJDR can aid this process by providing information for clinical and forensic staff to facilitate autopsy consent and referral across NSW and other states and territories. For all NSW and ACT cases, the department of forensic medicine at the Sydney LHD can perform autopsies, and the department of neuropathology at Royal Price Alfred Hospital, Sydney, can conduct neuropathological examination for diagnosis of CJD.
A new and promising diagnostic test called real-time quaking-induced conversion (RT-QUIC)24,25 can specifically detect and amplify the PrPsc protein from a sample of CSF. This antemortem test has the potential to improve the early diagnosis of CJD.
Although there are no effective treatments for CJD26, it is important to accurately identify and classify cases according to the diagnostic criteria discussed above, because of the invariably fatal nature of CJD and the potential for iatrogenic transmission.
Table 2. Tests currently used to diagnose and classify cases of Creutzfeldt–Jakob disease (CJD)
| Test | Use | Strengths | Limitations |
|---|---|---|---|
| Electroencephalogram (EEG)16,18 | Detects and measures the location and pattern of brain activity, which changes as the disease progresses | Periodic sharp-wave complexes (PSWC) occur in ~60% of sporadic CJD cases16 Sensitivity ranges from 12% to 72%, depending on molecular subtype18 |
Typical periodic EEG activity (i.e. PSWC) may be lacking in patients with sporadic CJD (in the very early and late stages of disease) |
| 14-3-3 protein in cerebrospinal fluid (CSF)17 | Detects the 14-3-3 protein in CSF, which is a nonspecific marker of central nervous system damage | Moderately accurate in diagnosing CJD Data from the Australian National CJD Register indicate sensitivity and specificity of ~88% |
Requires a lumbar puncture Nonspecific test, positive for other central nervous system pathology A positive result needs to be interpreted in the context of the pre-test probability of CJD |
| Magnetic resonance imaging (MRI)15,19 | Detects lesion patterns in the brain that are characteristic of prion protein accumulation and associated tissue damage | Cortical signal increase and hypersensitivities in basal ganglia and thalamus are characteristic across all subtypes of sporadic CJD Sensitivity of 58–71% and specificity of 82–90%19 |
MRI changes are frequent, differ across subtypes and should only be used as a surrogate indicator of CJD |
| Genetic analysis of prion protein gene (PRNP) gene20,21 | Detects polymorphisms and mutations in the PRNP gene | Helps diagnose individuals and families with genetic predispositions | Genome-wide association studies have indicated that other genes may contribute to the development of CJD and that these genes may not be tested for21 (e.g. ZBTB38–RASA2 has been implicated in the development of CJD cases in the UK) |
Figure 3. Age-standardised rates for classical Creutzfeldt–Jakob disease in NSW residents by Local Health District, 2004–13
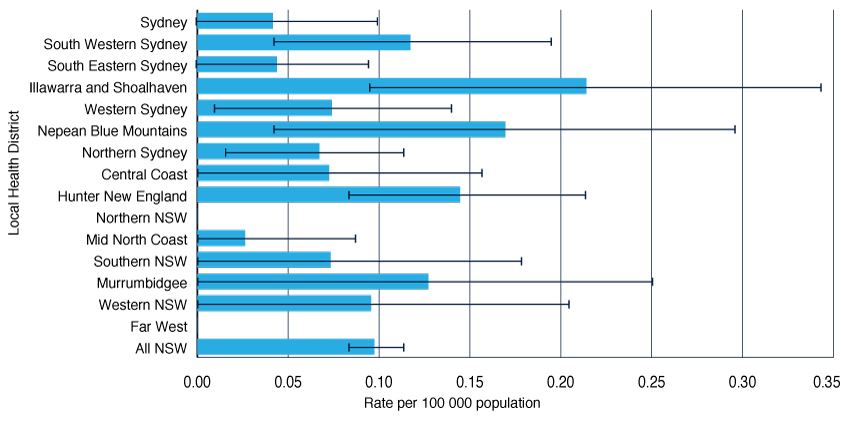
Note: Error bars show 95% confidence intervals
Prevention in the healthcare setting
The Communicable Diseases Network Australia has recently completed an updated version of its CJD infection control guidelines.27 Although CJD transmission events in the healthcare setting are rare28, these guidelines provide an evidence-based approach to managing any potential transmission risk associated with healthcare procedures.29 For example, any medical equipment used in a healthcare procedure on patients at increased risk of CJD, where the procedure involves high-infectivity tissues (e.g. brain, dura mater, spinal cord tissue), should be carefully incinerated or reprocessed according to the guidelines.27 Where possible, single-use equipment is preferred.
Conclusion
Although CJD is a rare disease in NSW and across Australia, evidence suggests that current routine surveillance systems for CJD may underestimate the true burden of disease.12 This highlights the importance of continued vigilance for the clinical signs of CJD, as well as recognition of the use and limitations of the different diagnostic tests available. Accurate diagnosis of CJD is important to reduce further transmission of CJD in the population. However, limiting the number of false positive results for this fatal condition is of paramount importance for patients and their families, and for others who may be affected by the public health actions that follow. In particular, clinicians and PHU staff should consider the pre-test probability of CJD when ordering and then interpreting a CSF 14-3-3 protein test. The Australian CJD infection control guidelines27 provide an evidence-based approach for clinical and PHU staff to be able to confidently assess and manage any potential transmission risk in the healthcare setting.
Acknowledgements
This work was completed while Emma Quinn was a trainee on the NSW Public Health Officer Training Program, funded by the NSW Ministry of Health. She undertook this work while based at the PHU in Tamworth, Hunter New England Population Health. The authors would like to thank Fakhrul Islam and Michelle Butler from Hunter New England Population Health for their assistance with the NCIMS data. The authors would also like to sincerely thank Genevieve Klug and Alison Boyd from the ANCJDR for their advice regarding the manuscript.
Copyright:

© Quinn et al. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International Licence, which allows others to redistribute, adapt and share this work non-commercially provided they attribute the work and any adapted version of it is distributed under the same Creative Commons licence terms.
References
- 1. Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559. Epub 2011 Dec 27. CrossRef | PubMed
- 2. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618–28. Epub 2012 Jun 20. CrossRef | PubMed
- 3. Wadsworth JD, Collinge J. Molecular pathology of human prion disease. Acta Neuropathol. 2011;121(1):69–77. Epub 2010 Aug 10. CrossRef | PubMed
- 4. Gambetti P, Cali I, Notari S, Kong Q, Zou WQ, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121(1):79–90. Epub 2010 Nov 09. CrossRef | PubMed
- 5. Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol. 2006;4(10):765–75. Epub 2006 Sep 19. CrossRef | PubMed
- 6. Allars M. Report of the inquiry into the use of pituitary derived hormones in Australia and Creutzfeldt-Jakob Disease. Canberra: Australian Government Publishing Service; 1994
- 7. Klug GM, Boyd A, McGlade A. Surveillance of Creutzfeld-Jakob disease in Australia: update to December 2012. Commun Dis Intell. 2013;37(2):E115–E20.
- 8. Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586–91. Epub 2005 May 11. CrossRef | PubMed
- 9. NSW Ministry of Health. Creutzfeld-Jakob disease: control guideline for public health units. Sydney: Communicable Diseases Branch, NSW Department of Health; 2004.
- 10. Klug GM, Boyd, A, McGlade,et al. Surveillance of Creutzfeld-Jakob disease in Australia: update to December 2011. Commun Dis Intell. 2012;36(2):E174–E9.
- 11. World Health Organization. Variant Creutzfeldt–Jacob disease. Fact sheet 180. Geneva: World Health Organization; 2012 [cited 2014 Sep 4]. Available from: https://www.who.int/mediacentre/factsheets/fs180/en/
- 12. Klug GM, Wand H, Boyd A, Law M, Whyte S, Kaldor J, et al. Enhanced geographically restricted surveillance simulates sporadic Creutzfeldt-Jakob disease cluster. Brain. 2009;132(Pt 2):493–501. Epub 2008 Dec 02. PubMed
- 13. Gelpi E, Heinzl H, Hoftberger R, Unterberger U, Strobel T, Voigtlander T, et al. Creutzfeldt-Jakob disease in Austria: an autopsy-controlled study. Neuroepidemiology. 2008;30(4):215–21. Epub 2008 Apr 22. CrossRef | PubMed
- 14. Klug GM, Wand H, Simpson M, Boyd A, Law M, Masters CL, et al. Intensity of human prion disease surveillance predicts observed disease incidence. J Neurol, Neurosur PS. 2013:84:12(1372–1377). Epub 2013 Aug 24. PubMed | Full text
- 15. Meissner B, Kallenberg K, Sanchez-Juan P, Collie D, Summers DM, Almonti S, et al. MRI lesion profiles in sporadic Creutzfeldt-Jakob disease. Neurology. 2009;72(23):1994–2001. Epub 2009 Jun10. CrossRef | PubMed
- 16. Wieser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol. 2006;117(5):935–51. Epub 2006 Jan 31. CrossRef | PubMed
- 17. Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2012;79(14):1499–506. Epub 2012 Sep 21. CrossRef | PubMed
- 18. Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129(Pt 9):2278–87. Epub 2006 Jul 04. CrossRef | PubMed
- 19. Tschampa HJ, Kallenberg K, Urbach H, Meissner B, Nicolay C, Kretzschmar HA, et al. MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease: a study on inter-observer agreement. Brain. 2005;128(Pt 9):2026–33. Epub 2005 Jun 17. CrossRef | PubMed
- 20. Windl O, Dempster M, Estibeiro JP, Lathe R, de Silva R, Esmonde T, et al. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum genet. 1996;98(3):259–64. Epub 1996 Sep 1. CrossRef | PubMed
- 21. Lloyd SE, Mead S, Collinge J. Genetics of prion diseases. Curr Opin Genet Dev. 2013;23(3):345–51. Epub 2013 Mar 23. CrossRef | PubMed
- 22. Hsich G, Kenney K, Gibbs CJ, et al. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. New Engl J Med. 1996;335(13):924–30. Epub 1996 Sep 26. CrossRef | PubMed
- 23. Collins S, Boyd A, Fletcher A, Gonzales M, McLean CA, Byron K, et al. Creutzfeldt-Jakob disease: diagnostic utility of 14-3-3 protein immunodetection in cerebrospinal fluid. J Clin Neurosci. 2000;7(3):203–8. Epub 2000 Jun 02. CrossRef | PubMed
- 24. Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17(2):175–8. Epub 2011 Feb 1. CrossRef | PubMed
- 25. Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, et al. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5(3):211–2. Epub 2008 Mar 01. CrossRef | PubMed
- 26. Stewart LA, Rydzewska LH, Keogh GF, Knight RS. Systematic review of therapeutic interventions in human prion disease. Neurology. 2008;70(15):1272–81. Epub 2008 Apr 09. CrossRef | PubMed
- 27. Communicable Disease Network Australia. Australian Creutzfeld-Jakob disease infection control guidelines. Canberra: Department of Health and Ageing; 2013.
- 28. Alcalde-Cabero E, Almazan-Isla J, Brandel JP, Breithaupt M, Catarino J, Collins S, et al. Health professions and risk of sporadic Creutzfeldt-Jakob disease, 1965 to 2010. Euro surveill. 2012;17(15). Epub 2012 Apr 21. PubMed | Full text
- 29. Koehler A, Athan, E, Collins, SJ. Updated Creutzfeld-Jakob disease infection control guidelines: sifting facts from fiction. Med J Australia. 2013;198(5):245. CrossRef | PubMed